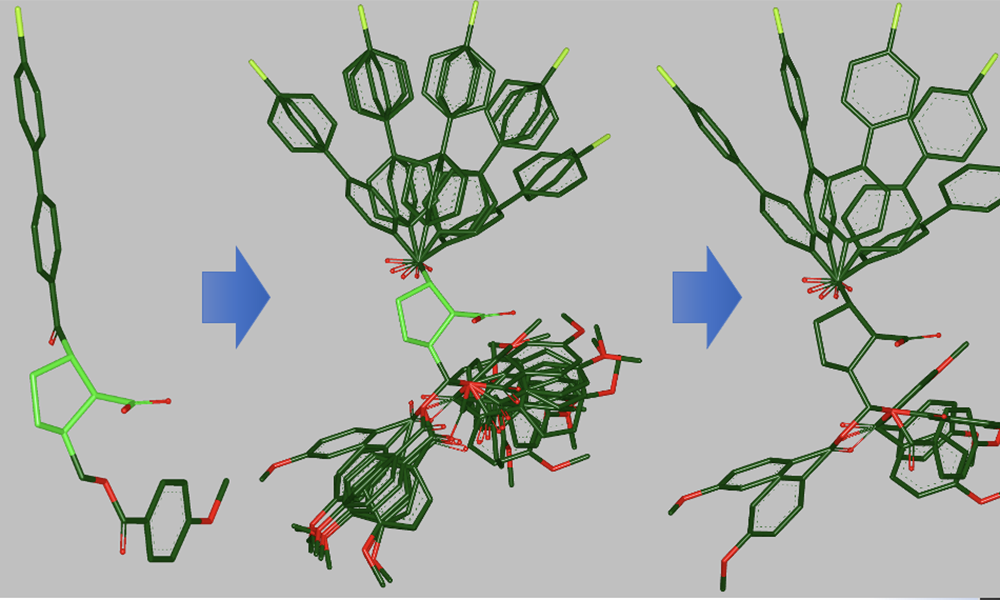
In the realm of drug discovery and molecular modeling, understanding the three-dimensional (3D) structure of molecules is crucial. Molecular conformers, which represent different spatial arrangements of atoms within a molecule, play a pivotal role in determining a molecule’s properties and interactions. In recent years, there have been significant advancements in the field of computational chemistry, and one groundbreaking approach is the generation of 3D molecular conformers through Equivariant Coarse-Graining and Aggregated Attention. In this blog post, we’ll delve into the details of this innovative technique and explore its implications for drug discovery and molecular science.
Understanding Molecular Conformers
Molecular conformers are distinct 3D arrangements of atoms within a molecule that result from rotations around single bonds. These conformers can significantly impact a molecule’s behavior, including its biological activity, stability, and reactivity. Thus, accurately predicting and generating molecular conformers is essential for various applications, such as drug design, virtual screening, and materials science.
Challenges in Molecular Conformer Generation
The generation of molecular conformers is a computationally intensive task due to the vast conformational space that molecules can explore. This challenge is exacerbated for larger and more complex molecules. Traditional methods, like molecular mechanics or molecular dynamics simulations, can be time-consuming and may not guarantee the discovery of all relevant conformers.
Equivariant Coarse-Graining and Aggregated Attention
Equivariant Coarse-Graining and Aggregated Attention is a cutting-edge approach that leverages deep learning and neural networks to predict 3D molecular conformers efficiently. Let’s break down the key components of this technique:
- Equivariant Coarse-Graining: Coarse-graining is a process that simplifies the representation of a system by reducing the number of degrees of freedom. In this context, equivariant coarse-graining is used to reduce the complexity of the molecule’s representation while preserving the essential structural information. This step helps in accelerating subsequent computations.
- Aggregated Attention: Attention mechanisms, which are prevalent in natural language processing, have found their way into the realm of computational chemistry. Aggregated Attention focuses on the important interactions between atoms and uses attention weights to prioritize interactions that significantly affect the molecule’s conformation.
Workflow of Equivariant Coarse-Graining and Aggregated Attention
Here’s a simplified workflow of how this approach generates 3D molecular conformers:
- Data Preparation: The first step involves collecting a dataset of molecules with known 3D conformers. These molecules are represented in a suitable format for deep learning.
- Equivariant Coarse-Graining: The molecule’s representation is coarsened, reducing the number of atoms and bonds while retaining crucial structural information.
- Aggregated Attention Network: A neural network model, equipped with aggregated attention layers, is trained on the coarsened representations of molecules. The attention mechanism allows the model to learn the significant interactions within the molecule.
- Conformer Generation: Once trained, the model can predict 3D conformers for a given molecule by leveraging its learned knowledge of important atomic interactions.
Advantages and Implications
The Equivariant Coarse-Graining and Aggregated Attention approach offers several advantages:
- Speed and Efficiency: By reducing the complexity of the molecular representation, this technique accelerates conformer generation, making it feasible for larger molecules and high-throughput screening.
- Accuracy: The use of attention mechanisms allows the model to capture the most critical atomic interactions, leading to more accurate conformer predictions.
- Drug Discovery: This approach has significant implications for drug discovery. It enables researchers to explore a broader range of molecular conformers, potentially identifying new drug candidates with improved properties.
- Materials Science: Beyond pharmaceuticals, this technique can be applied to materials science, helping researchers understand the properties and behavior of complex molecules in various materials.
The generation of 3D molecular conformers via Equivariant Coarse-Graining and Aggregated Attention represents a remarkable advancement in the field of computational chemistry. It combines the power of deep learning and attention mechanisms to accelerate and enhance conformer prediction, offering exciting possibilities for drug discovery and materials science. As this technology continues to evolve, it promises to reshape the way we approach molecular modeling and design, potentially leading to groundbreaking discoveries in chemistry and biology.
Certainly, let’s delve deeper into some of the technical aspects and potential applications of the Equivariant Coarse-Graining and Aggregated Attention approach for generating 3D molecular conformers.
Technical Details
- Representation Learning: The success of this approach heavily relies on the ability of the model to learn meaningful representations of molecules. This involves encoding molecular structures into vectors that capture essential information about atom types, bond connectivity, and spatial arrangements. Equivariant coarse-graining helps simplify these representations while retaining the critical information.
- Attention Mechanism: The use of an attention mechanism in the model is pivotal. It allows the network to focus on specific atomic interactions, such as steric clashes, hydrogen bonding, and van der Waals forces, that are vital in determining molecular conformations. The attention weights can be interpreted as the model’s learned understanding of the significance of different atomic interactions.
- Training Data: The quality and diversity of the training dataset are crucial. To ensure that the model generalizes well, it should be trained on a diverse set of molecules with known 3D conformers. Data augmentation techniques, such as rotating and translating molecules, can help increase the dataset’s diversity.
Applications
- Drug Discovery: The pharmaceutical industry stands to benefit significantly from this technology. Predicting accurate 3D molecular conformers allows researchers to perform virtual screening of vast chemical libraries, identifying potential drug candidates quickly and cost-effectively. This can expedite drug development and reduce the need for expensive and time-consuming laboratory experiments.
- Polymer Chemistry: Equivariant Coarse-Graining and Aggregated Attention can be applied to understand the conformations of complex polymer chains. This has implications in the design of new materials with specific properties, such as polymers with tailored mechanical or thermal characteristics.
- Catalysis Design: In catalysis, the arrangement of atoms at the active site of a catalyst is crucial. This approach can help design catalysts with optimal conformations for specific chemical reactions, potentially leading to more efficient and sustainable processes.
- Materials Discovery: Beyond polymers, this technology can assist in the discovery of novel materials with unique properties. For example, it can be used to predict the crystal structures of organic molecules, aiding in the development of advanced materials for electronics, energy storage, and more.
- Protein-Ligand Interactions: Understanding the 3D conformations of protein-ligand complexes is fundamental in drug discovery. This approach can be extended to predict ligand binding poses within protein binding sites, providing insights into drug-target interactions.
Challenges and Future Directions
- Scalability: While Equivariant Coarse-Graining and Aggregated Attention improve efficiency, handling extremely large molecules or systems with multiple interacting molecules remains a challenge. Future research may focus on optimizing scalability.
- Incorporating Solvation Effects: In real-world applications, molecules exist in a solvent environment, which can influence their conformations. Integrating solvation effects into the model is a complex but essential step for more accurate predictions.
- Interpretable Models: As with any deep learning approach, understanding why a model makes certain predictions is important. Developing methods to interpret the attention weights and learned representations can enhance the model’s transparency and trustworthiness.
In conclusion, the Equivariant Coarse-Graining and Aggregated Attention approach represents a remarkable fusion of deep learning and molecular science. Its potential impact on drug discovery, materials science, and various other fields is profound. As researchers continue to refine and expand upon this technique, we can expect it to play an increasingly pivotal role in accelerating scientific discoveries and technological advancements.
Read More : Google turns up the heat on AWS, claims Cloud Spanner is half the cost of DynamoDB


